COMBINE 2024

The “Computational Modeling in Biology” Network (COMBINE) is an initiative to coordinate the development of the various community standards and formats in systems biology and related fields. COMBINE 2024 will be a workshop-style event hosted at the University of Stuttgart in Stuttgart, Germany with support of the German Research Foundation (DFG) through the International scientific events program, the Stuttgart Center for Simulation Science (SimTech) and the Stuttgart Research Center Systems Biology (SRCSB). The meeting will be held in September 2024, closely aligned with the dates of the Virtual Physiological Human (VPH)2024 Conference September 04-06 Stuttgart, Germany.
The official programm takes place from Sunday 01st to Tuesday 3rd September. On Wednesday 4th and Thursday 5th September, there will be rooms provided for personal meetings only. The meeting days will include talks about the COMBINE standards and associated or related standardization efforts, presentations of tools using these standards, breakout sessions for detailed discussions as well as tutorials. There will be also a dedicated poster session, participants are encouraged to bring posters - poster boards will be provided next to meeting places. Some time each day will be left for community discussion and wrap-ups of breakouts and advertisements for following breakouts. It will be primarily an in-person meeting, with individual breakout sessions responsible for enabling remote participation as needed.
Local organizers are Nicole Radde (nicole.radde@simtech.uni-stuttgart.de) and Sebastian Höpfl (sebastian.hoepfl@isa.uni-stuttgart.de).
Confirmed Keynote Speakers and Session Chairs
- Irina Balaur, University of Luxembourg, Luxembourg
- Dirk Drasdo, Director of Research INRIA (French National Institut(ion) for Research in Computer Science and Control), Rocquencourt, France
- Fabian Fröhlich, Francis Crick Institute, United Kingdom
- Marc-Thorsten Hütt, Constructor University Bremen, Germany
- Matthias König, Humboldt-University Berlin, Germany
- Heinz Köppl, TU Darmstadt, Germany
- Göksel Misirli, Keele University, United Kingdom
- David Nickerson, Auckland Bioengineering Institute, University of Auckland, New Zealand
- Herbert Sauro, University of Washington, USA
- Lucian Smith, University of Washington, USA
- Judith Wodke, University Medicine Greifswald, Germany
Funded by
![]()
![]() and
and ![]()
A tentative schedule
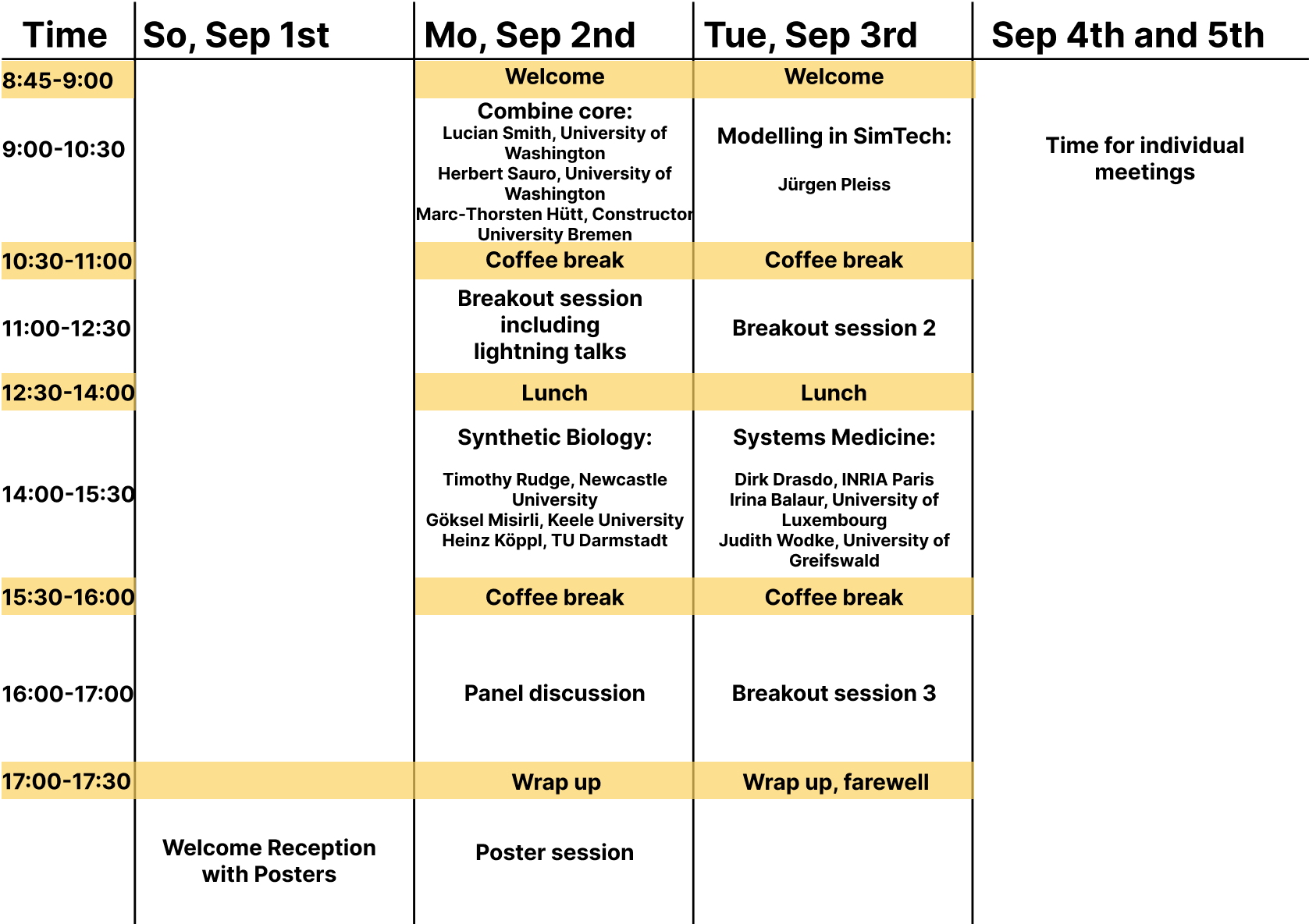 Note that many events are scheduled somewhat spontaneously at these events; keep an eye out here or on the COMBINE slack for last-minute changes and additions.
Note that many events are scheduled somewhat spontaneously at these events; keep an eye out here or on the COMBINE slack for last-minute changes and additions.
Workshop Location
COMBINE 2024 takes place at the University of Stuttgart in Stuttgart, Germany. COMBINE 2024 will take place in the former Campus Guest building, at Universitätsstrasse 34, 70569 Stuttgart. If you do not find the venue, email sebastian.hoepfl@isa.uni-stuttgart.de or nicole.radde@simtech.uni-stuttgart.de, or ping us on the COMBINE slack channel.
Arrival and Transportation
The closest airport is the Stuttgart Airport (STR). However, overseas the International Airport of Frankfurt Frankfurt Airport (FRA) is probably the better choice. There are ICEs and ICs going from the Frankfurt Airport to Stuttgart every one to two hours. You can book the train tickets via the Deutsche Bahn website. The Campus in Stuttgart, where we will host the Combine2024 is a 10 minute drive from the city center of Stuttgart. Unfortunately, the main line of the Sbahn is still closed during the COMBINE period. There is a rail replacement service (SEV) that serves all stops every 5 minutes Replacement Service. You can take any of these buses to the Universität stop when arriving at Stuttgart Central Station. If you arrive at Stuttgart airport, take the S-Bahn S2 to „Schorndorf“ or S3 to „Backnang“. In this case you can change to the bus replacement service (SEV) at the train station “Stuttgart Vaihingen”. Alternatively, if you come by car, set your destination at “Universitätsstrasse 34. 70569 Stuttgart (Campus Vaihingen)”.
Accomodation
There are no hotels at the workshop venue. However, you can find accommodations along the S-Bahn route, such as near the stations “Hauptbahnhof”, “Stadtmitte”, “Feuersee” or „Schwabstraße" (e.g. Studierendenhotel Stuttgart, Römerhof Vaihingen). From these stations you will need approximately 15-20 minutes to the campus.
Breakout sessions
 Detailed schedule for Breakout sessions including room numbers.
Detailed schedule for Breakout sessions including room numbers.
Training Models using PEtab Fröhlich, Fabian
PEtab is a standardized file format for specifying parameter estimation problems. The interoperable format is currently supported by 11 different tools, enabling users to benefit from standardized parameter estimation across frameworks based in Python, Julia, R, MATLAB, C++, or GUIs. Although PEtab was initially developed for parameter estimation, recent efforts have extended the format to improve standardization of various adjacent tasks, including: model selection, multi-scale modeling, PKPD and NLME modeling, optimal control, and visualization. In this breakout session, based on audience interests, we will present introductions to PEtab and its extensions, then discuss current efforts to improve PEtab. People unfamiliar with PEtab are welcome to attend, and might first like to check out the docs.
Introduction into Nix for scientific software Hauser, Simon
Working on software in a team brings all kinds of challenges, especially because everyone has a slightly different development environment.
These challenges usually start with onboarding new team members, include complications of moving your local environment to a high performance cluster and end in unreproducible bugs that boil down to ““works on my machine””.
Some of these issues can be resolved by providing dependency pinning using poetry or other package managers, but these solutions do not cover the operating system and require additional install documentation that usually contains apt usage.
Nix is a general purpose package manager that emerged in the last couple of years that solves these issues, by not just pinning the version of dependencies but also system libraries and tools, like the glibc library, python and also python packages.
This session will cover the fundamentals of Nix, including installation, command usage and writing your own custom development environment for a specific software.
Participants will learn how to leverage Nix to create reproducible scientific workflows, manage dependencies, and ensure consistent software environments across different systems.
Through practical demonstrations and hands-on activities, attendees will gain the skills necessary to integrate Nix into their scientific projects, enhancing both the reliability and portability of their software.
Join us to discover how Nix can streamline your scientific software development and deployment processes, fostering greater collaboration and innovation in your research endeavors.
A COMBINE Standard for Multi-Approach Multi-Scale (MAMS) Modelling Sheriff, Rahuman
Multi-approach Multi-scale (MAMS) modelling represents a cutting-edge method for modelling and analysis of biological systems, leveraging an integrated suite of diverse modelling frameworks. This multi-approach modelling will encompass a combination of diverse modelling formalisms, such as ordinary differential equations (ODE), partial differential equations (PDE), logical, constraint-based, and agent-based models across multiple scales. These models are intricately tied together to facilitate complex simulations. During the dedicated breakout sessions at Harmony 2021, COMBINE 2021, and HARMONY 2024, we delved into the existing state-of-the-art technologies and standards, including SBML and SED-ML, and their support for multi-approach modelling. These discussions also illuminated the current challenges and gaps within the field. For COMBINE 2024, our objective is to further this conversation by identifying published MAMS models and bringing together the community to enable the creation of novel standards or the enhancement of existing COMBINE standards to support MAMS. This effort aims to foster interoperability and support the rapidly evolving paradigm of MAMS modelling.
Continuing Work Towards Reproducible Stochastic Biological Simulation Sego, T.J.
Stochastic simulations are commonly used to quantitatively or semi-quantitatively describe the dynamics of biological systems. At various scales and in multiple applications, stochastic simulation better reflects observed biological processes and robustness. Various methods are widely used to incorporate stochasticity into biological simulation, such as the Gillespie stochastic simulation algorithm for systems biology modeling, stochastic Boolean networks for network modeling, and the Cellular Potts model methodology for multicellular modeling. Proving reproducibility of simulation results is critical to establishing the credibility of a model. To this end, BioModels, the largest repository of curated mathematical models, tests and reports the reproducibility of simulation results for all submitted models when possible. A recent study showed that about 50% of the deterministic ordinary differential equation models on BioModels could not be reproduced when applying criteria for reproducibility to the information provided in their associated publication, reflecting a current crisis of reproducibility. Furthermore, there are no well-accepted metrics or standards for reproducing stochastic simulation results, thus perpetuating the crisis of reproducibility for a broad class of biological models. This breakout session will continue work towards establishing an accepted framework for testing the reproducibility of stochastic simulations in biological modeling. The session will provide a brief overview of recent progress towards defining quantitative measures to determine whether stochastic simulation results can be reproduced, and when results have been reproduced. Attendees will discuss current issues to address towards consensus and broad adoption in relevant modeling communities, as well as future work towards reproducibility of stochastic simulation results using multiscale and complex models.
SBGN PD: current and future development Rougny Adrien
Visualization of biological processes plays an essential role in life science research. Over time, diverse forms of diagrammatic representations, akin to circuit diagrams, have evolved without well-defined semantics potentially leading to ambiguous network interpretations and difficult programmatic processing. The Systems Biology Graphical Notation (SBGN) standard aims to reduce ambiguity in the visual representation of biomolecular networks. It provides specific sets of well-defined symbols for various types of biological concepts. SBGN comprises three complementary languages: Process Description (PD), Entity Relationship (ER), and Activity Flow (AF). The XML-based SBGN Markup Language (SBGN-ML) facilitates convenient storage and exchange of SBGN maps. The SBGN languages as well as SBGN-ML are described in detail in specifications (see sbgn.org). This breakout session will focus on the development of SBGN PD. We invite all participants interested in SBGN to join this session, where we will discuss specific issues related to the next version of the PD specification, as well as more open issues related to a future level of SBGN PD.
Combine spatial multi-cellular modelling with SBML Jörn Starruß
Modularity is key to creating complex multi-cellular models while preserving the accessibility of meaningful submodels. Naturally, composition also encourages reusability and the likes. We want to discuss and establish a common practice how to overlay the spatial dynamics of multi-cellular models with reaction dynamics defined in the SBML standard.
Most obvious features to be represented separately from the spatial cell dynamics are intra-cellular regulatory systems, inter-cellular communication and spatial reaction-diffusion processes using SBML-spatial. Further issues arise when inter-connecting identical submodels residing in individual cells and the definition of instantaneous assignments upon entity operations (e.g. cell birth & death).
As an introductory motivation we will present our latest Morpheus results in embedding spatial reaction-diffusion submodels within moving cells. Using that experience we will sketch a way how to exploit the HMC package to compose SBML models and attach them in a second step to the individual scopes of our spatial model. We hope for a lively discussion on best practice approaches interconnecting spatial multi-cellular modeling and the SBML standard.
OpenVT–Developing Framework Description Standards for MultiCellular Agent-Based Virtual Tissue Models Glazier, James
Many simulation frameworks implement multicellular agent-based models using a variety of methodologies (center model, vertex model, Cellular Potts model, finite-element mechanics,….) and support a variety of biological and mathematical processes it can be often confusing and time consuming to for a researcher to know which simulation framework can fulfill their modeling needs. This session will discuss an approach to defining and categorizing simulation framework capabilities. The session will start with an overview of the various methods employed in multicellular simulations, highlighting their unique features and common challenges. It will present our approach to describing framework descriptions in a standardized way followed by discussion on this approach.
OpenVT–MultiCellular Agent-Based Virtual Tissue Models: Defining Topics and Priorities Glazier, James
Virtual Tissues (VT), agent based multicellular modeling has become indispensable in understanding complex biological phenomena, from tissue development to disease progression. But the diversity in simulation methods poses challenges in reproducibility, modularity, reusability, and integration for multiscale models, leading to a fragmented ecosystem and hindering growth. The OpenVT Community is trying to address these challenges by bringing siloed research groups together to improve the sharing of VT knowledge. The OpenVT Community supports the expansion of and broader adoption of multicellular modeling beyond academic research labs into greater industry practice. Development of best practices and better reproducibility will ultimately lead to models that more closely follow FAIR (Findable, Accessible, Interoperable, and Reusable) principles, leading to wider use in therapeutic approaches, toxicology, drug discovery and personalization of testing and treatment. This session aims to discuss current progress undertaken by the OpenVT community towards a shared ecosystem and look to attendees for insight into what they believe will encourage broader adoption of community guidelines.
OpenVT–Developing Reference Models for Multicellular Agent-Based Virtual Tissue Models Glazier, James
An increasing number of packages implement multicellular agent-based models using a variety of methodologies (center model, vertex model, Cellular Potts model, finite-element mechanics,….). In principle a set of underlying biological and physical processes should yield the same result independent of the package in which they are implemented. However, at the moment, comparison between methodologies or even between different packages implementing the same methodology, is quite challenging. As a first step to building a shared understanding of modeling capabilities and to improve rigor and reproducibility, we define a minimal set of standard reference models which should be implemented in each framework to illustrate their capabilities and reveal hidden discrepancies of approach. This session will discuss these efforts and look for feedback from attendees.
Tutorials
Workshop: refineGEMs and SPECIMEN for automated model reconstruction and annotations Döbel, Gwendolyn O.
Metabolic model reconstruction usually relies on several cumbersome steps. Different tools exist, which are only partially automated and need to be connected manually. Our aim is to simplify and reduce the manual workload. Thus, we developed the toolbox refineGEMs and the workflow collection SPECIMEN. A stable release of refineGEMs was already used in practice. Both tools are currently under active development (enhancement and extension).
This workshop aims to give the attendees a brief introduction to automatic metabolic modelling with the tools refineGEMs and SPECIMEN. As part of the workshop, an open discussion will be held about issues arising from automatic energy-generating cycle (EGC) dissolution and gap filling.
Biological and Biophysics Simulation in Tissue Forge: Introduction and Guided Simulation Building Sego, T.J.
Tissue Forge is open-source simulation software for interactive particle-based physics, chemistry and biology modeling and simulation. Tissue Forge allows users to create, simulate and explore models and virtual experiments based on soft condensed matter physics at multiple scales, from the molecular to the multicellular, using a simple interface. While Tissue Forge is designed to simplify solving problems in complex subcellular, cellular and tissue biophysics, it supports applications ranging from classic molecular dynamics to agent-based multicellular systems with dynamic populations. Tissue Forge users can build and interact with models and simulations in real-time and change simulation details during execution, or execute simulations off-screen and/or remotely in high-performance computing environments. Tissue Forge provides a growing library of built-in model components along with support for user-specified models during the development and application of custom, agent-based models. Tissue Forge includes an extensive Python API for model and simulation specification via Python scripts, an IPython console and a Jupyter Notebook, as well as C and C++ APIs for integrated applications with other software tools. Tissue Forge supports installations on Windows, Linux and MacOS systems and is available for local installation via conda. This tutorial introduces the basic concepts, modeling and simulation features, and some relevant modeling applications of Tissue Forge through guided simulation scripting. Tutorial concepts will introduce basic Tissue Forge modeling concepts and simulation features through the development of interactive simulations in Python. Attendees are encouraged, but not required, to code along as the tutorial interactively develops and tests simulations in multicellular and biophysics modeling applications.
Morpheus: A user-friendly simulation framework for multi-cellular systems biology Starruß, Jörn and Brusch, Lutz
“Multi-cellular modeling and simulation become increasingly important to study tissue morphogenesis and disease processes. This tutorial introduces Morpheus in an overview presentation with live demos and hands-on exercises runnable in sync on the presenter’s and your own laptop. The focus lies on importing SBML models into Morpheus, extending them in space as reaction-diffusion processes and automatically ““cloning”” them into many individual cells that can dynamically interact. Also, own modeling ideas can be explored with the help of a tutor. Morpheus offers modeling and simulation of multi-cellular dynamics in a Graphical User Interface (GUI) without the need to program code. It uses the domain-specific language MorpheusML to define and simulate multicellular models in 3D space including the most common cell behaviors and tissue mechanics. Morpheus is open-source software and provides readily installable packages for macOS, Windows, Linux. Please download before the tutorial and have a look around the homepage incl. >90 example models.
Talks
Results and Lessons learnt from the community-level project on “Fostering the uptake of RDA indicators in Systems Biomedicine as a measure for model quality and FAIRness within the COMBINE community” Irina Balaur
Biosimulation models incorporating multi-layered information have been developed to explore complex disease mechanisms. While these models have potential for diagnosis and therapy support, their clinical use is limited by unclear specifications on settings and kinetics, hampering reusability and reproducibility. Adhering to the principles of Findability, Accessibility, Interoperability, and Reusability (FAIR) can enhance the credibility and reproducibility of these models.
In this talk, we will focus on the results and lessons learned from our project on applying RDA indicators for the FAIR evaluation of biosimulation models within the COMBINE community . Funded by the EOSC Future, the project focused on developing community-level guidelines for FAIR model design and a semi-automatic FAIR evaluation tool for COMBINE models. We adapted the RDA FAIR indicators template from the IMI FAIRplus project to cover the specificities of the COMBINE computational simulations. The semi-automatic FAIR assessment tool enables some RDA FAIR indicators to be filled automatically.
We will also discuss the major challenges and strategies for achieving a community-accepted set of FAIR indicators specific to COMBINE resources. To address these challenges, we organized workshops and training sessions within the RDA and COMBINE communities, and engaged in transparent communication, online dissemination, and involvement acknowledgment. All components of our work are open, freely accessible, and connected via our project website to major frameworks like Zenodo for dissemination materials and GitHub for code files.
We also collaborated with other relevant projects on FAIR data and infrastructure. We will conclude by exemplifying our work on the FAIR assessment of the MINERVA platform, supporting the Disease Maps Project, and the ongoing development of a FAIR-calibration framework for reporting guidelines of AI-related studies in healthcare to enhance reproducibility and open science.
CompuTiX: A library for agent based modeling (not only) at a tissue-scale Jiří Pešek and Dirk Drasdo
In recent years, many studies have shown that the tissue microarchitecture along with the mechanical environment has a crucial yet poorly understood impact on the biological processes inside living tissues This have a significant impact on progression of any potential disease or treatment. The limitations of in-vivo imaging techniques together with the small scale and isolated nature of many in-vitro experiments, makes these systems a suitable candidate for in-silico approach, where initial in-vitro experiments can be used to formulate and tune the underlying models and in-vivo imaging is then used to generate a patient specific setup. In particular, an agent based models, where the global effect is achieved by interaction between many, relatively simple, entities, are suitable to capture the spatial and behavioral heterogeneity and complexity of living tissues. In this talk we will present a new open-source computational library, CompuTiX, suitable for agent based simulations of tissues, organoids and more. We will split the talk into two parts. In the first part we will briefly introduce basic bio-physical models starting from simple center based models to more complex models like deformable cell model. In the second, more technical, part we will discuss the architecture of the library, design choices, trade-offs and challenges in our goal to provide a versatile and extensible platform for agent based simulations.
Networks, simple models and model diversity in the description of biological systems Marc-Thorsten Hütt
“My talk will address three distinct, but interrelated, topics: (1) networks as structural models to interpret high-throughput data; (2) the distinction between mathematical models and their computer implementations; (3) simple models vs. complicated, parameter-rich models.
Systems biology and systems medicine frequently use network-based strategies for data interpretation and data contextualization. These methods, at times, lack standardization and comparability. Here I briefly discuss, how such methods work, and which implicit hypotheses are associated with them.
The formal representation of a mathematical model is often incomplete, compared to the details required for an implementation of the model to run numerical simulations. Implementation differences can in principle lead to drastically different results. For the case of models of excitable dynamics, I illustrate this point, showing that even the simplest models can display such implementation differences.
Lastly, residing on the topic of simple models, I briefly draw the attention to the co-existence of parameter-rich and simple models of biological systems, outlining a few pros and cons and caveats. "
Energy and Information in Gene Regulation Heinz Köppl
The essence of cellular function is to maintain a non-equilibrium state through an influx of chemical energy. In particular gene regulation involves several active steps that require energy carriers such as ATP. Thermodynamic uncertainty relations tell us that reducing fluctuations in molecular systems comes at the expense of energy consumption which in turn is related to the rate of entropy production. In this talk I will make this notion more concrete for Markov models of chemical reaction networks and in particular for simple models of gene expression. We will also introduce an operational notion of precision for a molecular signal processing systems using information theory. We show how entropy production rates provides bounds on the achievable information throughput for simple gene expression channels. We also discuss how this analysis can help in designing regulatory circuits in synthetic biology that are energy efficient and yet precise.
Computational design of biological receivers using multi-scale models and data standards Göksel Misirli
Engineering genetic regulatory circuits that sense external molecules and respond is essential for developing diverse biological applications. As the complexity of designs increases, a model-driven design process becomes desirable to explore large design spaces that involve different biological parts and parameters. Moreover, the amount of these molecules reaching a receiver is usually assumed to be constant, and the diffusion dynamics and the interference caused by late-arriving molecules and the cellular dynamics are often not integrated. Additionally, each molecule type may represent a single biological signal and be unsuitable for encoding and decoding multiple data bits. Here, we present the virtual parts repository, a computational framework that provides modular, reusable and composable models. The framework facilitates automating the design of predictable applications via simulations. It builds on the Systems Biology Markup Language to model cellular behaviour and the Synthetic Biology Open Language to capture the details of genetic circuits. We then extend this automation approach to design the end-to-end transmission of signalling molecules from a transmitter to cellular receivers for multi-bit data communications. The resulting framework can be used to understand the cellular response for a sequence of custom data bits, each representing a group of molecules released from a transmitter and diffusing over a molecular channel. The framework validates and verifies various communication parameters and identifies the best communication scenarios. We also present a novel algorithm to minimise signal interference by employing equalisation techniques from communication theory. Our data standards-enabled and multi-scale modelling workflow combines engineering genetic circuits and molecular diffusion dynamics to encode and decode data bits, design efficient cellular signals, minimise noise, and develop biologically plausible applications.
The past, present and possible futures Herbert Sauro
It has almost been 25 years since Hiroaki Kitano initiated the development of SBML as part of the ERATO project. Together with Bolouri, Doyle, Finney, Hucka, myself and a number of key stakeholders (who continue to meet at COMBINE), we published the first draft and software support libraries for the SBML specification. Around the same time we also saw the publication of the specification for CellML that was a more mathematically oriented proposal. What resulted was most unexpected, the emergence of a new vibrant ecosystem which stimulated further development, created a host of new ancillary standards as well as the indispensable BioModels repository. That ecosystem still exists today. In this talk I will review what I feel remains to be done or is incomplete, what new modeling challenges we face, and describe what the center of model reproducibility in the US is doing in terms of software provision. In particular I will describe a number of new client-based web tools and desktop apps. The client-based tools are unusual in that they can be hosted from any free basic server such as a GitHub, Neocities or Cloudflare page. This makes such apps very low maintenance and tend to persist long after funding stops. Examples from our center include a model annotation (AWE) platform, a simple model checking app (ratesb), a high speed BioModels cache, a reproducibility portal, a model verification service, a new SBML/Antimony web utility, a Biosimulators/Biosimulations repository, a new SBML compliant desktop app, a number of new python packages for network visualization, a new desktop network editor (Alcuin), new extensions to Antimony (See talk by Lucian Smith), a standard protocol for multi-scale modeling (See talk by Eran Agmon), and the first model credibility hackathon held this summer.
Improving Curation: Biomodels and Annotation Lucian Smith
The BioModels Database has over 1000 curated models from published papers. Curators at the EBI ensure that the model can be used to reproduce at least one figure from the paper, and extensively annotate the model as well. However, until the advent of SED-ML, it was impossible to store what the curator did to reproduce the model in a standard format, and until more widespread use of SED-ML, it was impossible to reliably validate any SED-ML that was produced. The Center for Reproducible Biomedical Modeling has produced new SED-ML interpreters and validators that have bridged this gap, and we have partnered with the EBI to ‘retro-curate’, as far as possible, the curated branch of BioModels, to include validated SED-ML, which we have then tested using the SED-ML interpreters on multiple simulation engines.
In addition, we have extended the Antimony modeling language, and present the Antimony Web Editor, with particular features useful for adding curation of species, reactions, and parameters.
MeDaX – two years towards bioMedical Data eXploration Judith Wodke
Research based on clinical care data is gaining attention across the world. However, the quality of clinical care data is generally not maximised for research purposes. Instead, according to economic principles, medical staff and time costs are commonly minimised, rendering the enrichment with sufficient metadata for easy data reuse at least challenging. In addition, a heterogeneous landscape of laws concerning medical data reuse on national, state, and county levels make (international) interoperability an ambitious aim. The MeDaX project was initiated about two years ago and its underlying idea presented at COMBINE 2022: connect and semantically enrich highly diverse clinical and other biomedical data in knowledge graphs (KG) to design, implement, and use graph technologies for innovative data exploration.
The MeDaX-KG prototype has been designed and implemented building on the BioCypher framework to harmonise biomedical knowledge graphs and using synthetic patient data. The proof of concept pipeline consists of i) a FHIR input adapter, including an optimisation module for the generically generated graph structure, ii) a semi-automatic data schema generation based on the BioLink ontology, and iii) the visualisation of the resulting MeDaX-KG using Neo4j. Currently, the pipeline is improved, a user interface is implemented, and the first pilot in a german university clinic’s data integration center is set up while the first stable release is prepared.
The EnzymeML framework: improving efficiency and quality of biocatalytic science Jürgen Pleiss
Biocatalysis is entering a promising era as a data-driven science. High-throughput experimentation generates a rapidly increasing stream of biocatalytic data, which is the raw material for mechanistic and data-driven modeling to design improved biocatalysts and bioprocesses. However, data management has become a bottleneck to progress in biocatalysis. In order to take full advantage of rapid progress in experimental and computational technologies, biocatalytic data should be findable, accessible, interoperable, and reusable (FAIR).
The EnzymeML framework provides reusable and extensible tools and a standardized data exchange format for FAIR and scalable data management in biocatalysis 1. To enable storage, retrieval, and exchange of enzymatic data, the XML–based markup language EnzymeML has been developed 2. An EnzymeML document contains information about reaction conditions and the measured time course of substrate or product concentrations. Kinetic modelling is performed by uploading EnzymeML documents to the modelling platforms COPASI or PySCeS or by using the JAX platform. The rate equation and the estimated kinetic parameters are then added to the EnzymeML document. The EnzymeML document containing the experimental and the modelling results is then uploaded to a Dataverse installation or to the reaction kinetics database SABIO-RK. The workflow of a project is encoded as Jupyter Notebook, which can be re-used, modified, or extended The feasibility and usefulness of the EnzymeML toolbox was demonstrated in six scenarios, where data and metadata of different enzymatic reactions are collected, analysed, and uploaded to public data repositories for future re-use 3.
FAIRification of data and software and the digitalization of biocatalysis improve the efficiency of research by automation and guarantee the quality of biocatalytic science by reproducibility4. Most of all, they foster reasoning and creating hypotheses by enabling the reanalysis of previously published data, and thus promote disruptive research and innovation.
preCICE – A General-Purpose Simulation Coupling Library Benjamin Ückermann
preCICE is an open-source coupling software for partitioned multi-physics and multi-scale simulations including PDE-PDE and PDE-ODE coupling. Thanks to the software’s library approach (the simulations call the coupling) and its high-level API, only minimally-invasive changes are required to prepare an existing (legacy) simulation software for coupling. Moreover, ready-to-use adapters for many popular simulation software packages are available, e.g. for OpenFOAM, SU2, CalculiX, FEniCS, and deal.II. For the actual coupling, preCICE offers methods for fixed-point acceleration (quasi-Newton acceleration), fully parallel communication (MPI or TCP/IP), data mapping (radial-basis function interpolation), and time interpolation (waveform relaxation). Today, although being an academic software project at heart, preCICE is used by more than 100 research groups in both academia and industry. In this presentation, I introduce the basic concepts of preCICE and discuss existing and potential applications in biology.
Reproducible tools for dealing with highly variable data Nicole Radde
In the biomedical context, data is often sparse, and replicates show a high variability. This is because complex procedures, costs, and ethical aspects constrain measurements. Sparsity and high variability pose a challenge for modeling, especially when building models aiming to capture quantitatively dynamic responses.
Here, we present two complementary approaches we developed in our group to deal with sparse and variable data. Bayesian Modeling of Time Series Data (BayModTS) uses a Bayesian approach and a simulation model to process sparse and highly variable serial data. BayModTS can be used to quantify uncertainty in the observed process or as a noise filtering approach, as we will demonstrate with selected examples.
Second, Eulerian Parameter Inference (EPI) formulates the parameter estimation problem for a simulation model from experimental data as a stochastic inverse problem and infers a parameter distribution that can reproduce the variability of the input data.
Both approaches are implemented as documented software packages that use standards such as SBML or PEtab. In my talk, I will briefly explain our methods and discuss the current challenges regarding reproducibility and FAIR principles from a modeler’s perspective.
Lightning talks
Modeling and simulation using industrial standards Modelica, FMI and web components. - Lightning talk session 2 Tomas Kulhanek
We use industrial standard Modelica to express complex models of human physiology [1,2]. Recently we have published enabling technology that allows to export complex models in standard functional mockup interface API (FMI) as a web component to be integrated with other web standards and technologies to create modern web application. Thanks to it the models does not necessarry need to be implemented in Modelica language, but and standard FMI needs to be implemented by other standards to compute model derivatives and do simulation step using a prefered numerical method.
Utilizing Nix for rapid BayModTS development - Lightning talk session 3 Hauser, Simon
BayModTS, a python project for FAIR Bayesian Modelling of Time Series workflows, has a complex setup that requires users of that software to install and compile multiple python packages that have native C dependencies.
This is a complex endeavor and currently it is not possible to fully resolve these issues using poetry install.
We present a solution that utilizes a general purpose package manager called Nix, that guarantees that a package and all its dependencies can be built reproducibly.
This package manager can be used to build all kinds of software packages, including C libraries and python packages, which we need to realize our solution.
Biological and Biophysics Simulation in Tissue Forge - Lightning talk session 4 Sego, T.J.
Tissue Forge is open-source simulation software for interactive particle-based physics, chemistry and biology modeling and simulation. Tissue Forge allows users to create, simulate and explore models and virtual experiments based on soft condensed matter physics at multiple scales, from the molecular to the multicellular, using a simple interface. While Tissue Forge is designed to simplify solving problems in complex subcellular, cellular and tissue biophysics, it supports applications ranging from classic molecular dynamics to agent-based multicellular systems with dynamic populations.
A COMBINE Standard for Multi-Approach Multi-Scale (MAMS) Modelling - Lightning talk session 1 Sheriff, Rahuman
Multi-approach Multi-scale (MAMS) modelling represents a cutting-edge method for modelling and analysis of biological systems, leveraging an integrated suite of diverse modelling frameworks. This multi-approach modelling will encompass a combination of diverse modelling formalisms, such as ordinary differential equations (ODE), partial differential equations (PDE), logical, constraint-based, and agent-based models across multiple scales. These models are intricately tied together to facilitate complex simulations.
Continuing Work Towards Reproducible Stochastic Biological Simulation - Lightning talk session 3 Sego, T.J.
Stochastic simulations are commonly used to quantitatively or semi-quantitatively describe the dynamics of biological systems. At various scales and in multiple applications, stochastic simulation better reflects observed biological processes and robustness. Various methods are widely used to incorporate stochasticity into biological simulation, such as the Gillespie stochastic simulation algorithm for systems biology modeling, stochastic Boolean networks for network modeling, and the Cellular Potts model methodology for multicellular modeling.
Morpheus model repository: Experiences with reproducible multi-cellular models - Lightning talk session 1 Brusch, Lutz
“Collaborative modeling and simulation become increasingly important for studying self-oganization, patterning, morphogenesis and disease processes from the intracellular to the tissue and organ scales. To support collaborations, we have developed the Morpheus model repository. This model repository is an open access and citable platform for publishing, sharing and archiving multi-scale and multi-cellular models that are encoded in the model description language MorpheusML.
Computational Model Development Using SBML: sbmlutils, sbm4humans, cy3sbml - Lightning talk session 4 König, Matthias
“The Systems Biology Markup Language (SBML)(doi 10.15252/msb.20199110) is recognized as the standard framework for representing and exchanging complex mathematical models in biological systems research. One of the primary challenges faced by newcomers in computational biology is the encoding and development of ordinary differential equation (ODE) models within the SBML framework. Addressing this hurdle, we introduce two innovative Python tools: sbmlutils, sbml4humans, and the Cytoscape application cy3sbml. These tools collectively streamline the process of SBML model creation, enhancing both the programmatic aspect and the user experience.
OpenVT–Developing Framework Description Standards for MultiCellular Agent-Based Virtual Tissue Models - Lightning talk session 3 Glazier, James
Many simulation frameworks implement multicellular agent-based models using a variety of methodologies (center model, vertex model, Cellular Potts model, finite-element mechanics,….) and support a variety of biological and mathematical processes it can be often confusing and time consuming to for a researcher to know which simulation framework can fulfill their modeling needs. In our breakout session, we will discuss an approach to defining and categorizing simulation framework capabilities. (Zoom link; was iu.zoom.us/meeting/register/tZUlcu6hpjoiG9OLZ1rGcrOfWkf49p4uVtjo)
OpenVT:MultiCellular Agent-Based Virtual Tissue Models: Defining Topics and Priorities for Working Groups and Virtual Workshops - Lightning talk session 1 Glazier, James
“Virtual Tissues (VT), agent based multicellular modeling has become indispensable in
understanding complex biological phenomena, from tissue development to disease progression. But the diversity in simulation methods poses challenges in reproducibility, modularity, reusability, and integration for multiscale models, leading to a fragmented ecosystem and hindering growth. The OpenVT Community is trying to address these challenges by bringing siloed research groups together to improve the sharing of VT knowledge. (Zoom link; was iu.zoom.us/meeting/register/tZUlcu6hpjoiG9OLZ1rGcrOfWkf49p4uVtjo)
OpenVT–Developing Reference Models for Multicellular Agent-Based Virtual Tissue Models - Lightning talk session 4 Glazier, James
An increasing number of packages implement multicellular agent-based models using a variety of methodologies (center model, vertex model, Cellular Potts model, finite-element mechanics,….). In principle a set of underlying biological and physical processes should yield the same result independent of the package in which they are implemented. However, at the moment, comparison between methodologies or even between different packages implementing the same methodology, is quite challenging. As a first step to building a shared understanding of modeling capabilities and to improve rigor and reproducibility, we define a minimal set of standard reference models which should be implemented in each framework to illustrate their capabilities and reveal hidden discrepancies of approach. (Zoom link; was iu.zoom.us/meeting/register/tZUlcu6hpjoiG9OLZ1rGcrOfWkf49p4uVtjo)
A functional tissue unit approach to understanding lung function in health and disease - Lightning talk session 2 Li, Ruobing
The primary functional tissue unit of the lungs is the acinus. An acinar unit brings together diverse functions, including airflow, blood flow, gas exchange, mechanical deformation and the effect of surfactant on this, and fluid transport from the blood to the lymphatic vessels. Existing models of varying geometric complexity have been developed to simulate lung mechanical behaviours and various fluid transport, currently as separate systems. This study addresses this gap by developing a respiratory FTU that integrates these different models to simulate acinar function and link this to represent whole lung function. Model implemented by CellML, Fortran, and Python. By integrating these individual models, we aim to provide a better understanding of the interactions and dependencies within the lungs, essential for simulating lung function in health and disease.
Updates to the Systems Biology Graphical Notation: A Standardized Representation of Biological Maps - Lightning talk session 1 Luna, Augustin
Visualization of biological processes plays an essential role in life science research. Over time, diverse forms of diagrammatic representations, akin to circuit diagrams, have evolved without well-defined semantics potentially leading to ambiguous network interpretations and difficult programmatic processing. The Systems Biology Graphical Notation (SBGN) standard aims to reduce ambiguity in the visual representation of biomolecular networks.
A Standardized Protocol for Integrative, Multiscale Modeling - Lightning talk session 1 Moraru, Ion
We are developing a standardized protocol for multi-algorithmic model composition, based on standardized schemas for process interfaces, composition patterns, and orchestration patterns. This will provide the foundation for robust infrastructure for systems biology models. The BioSimulators project aims to establish this protocol, ensuring reproducibility, tool compatibility, and “plug-and-play” integration of new processes and data.
Posters
GEMsembler: a package for comparing and combining ensembles of genome-scale metabolic models to study microbial metabolism Matveishina, Elena
SPECIMEN: Collection of Workflows for Automated and Standardised Reconstruction of Genome-Scale Metabolic Models Brune, Carolin
What is an appropriate standard for modeling microbial communities? Ruth, Beatrice
Shining Light on Single-Cell Dynamics and Heterogeneity: Design and analysis of a hybrid population model for an epigenetic memory system. Klingel, Viviane
TFpredict Molnár, Dóra Viktória
A Computational Pipeline for Evaluating Agreement Between Large-Scale Models and Diverse Datasets Huggins, Jonah
CellDesigner5: Biochemical network editor for large-scale modeling. Araki, Taichi
Integrate modelling standards with Energy-based System Analysis Ai, Weiwei
MomaPy: a Python library to work with molecular maps programmatically Rougny, Adrien
The preCICE v3 coupling library and the emerging preCICE ecosystem Chourdakis, Gerasimos
ModelPolisher: Enhancing the Quality and Completeness of Genome-Scale Metabolic Models (GEMs) Eltzner, Dario
Partitioned simulations using the neuromuscular simulation framework OpenDiHu Homs-Pons, Carme
SBSCL: A Library of Efficient Java Solvers and Numerical Methods to Analyze Computational Models in Systems Biology Neumann, Arthur
BayModTS: A Bayesian workflow to process variable and sparse time series data. Höpfl, Sebastian
Standard compliant data and model management for systems medicine projects Olga Krebs
The role of standards in defining an ecosystem for Virtual Human Twins (VHTs) Mayer, Gerhard
Recommendations and requirements for implementing computational models in clinical integrated decision support systems Golebiewski, Martin